Atomare und elektronische Eigenschaften von Festkörpergrenzflächen
Forschungsbericht (importiert) 2005 - Max-Planck-Institut für Festkörperforschung
Die Untersuchung von Oberflächen, Grenzflächen und dünnen Schichten stellt einen wichtigen Aspekt der Festkörperforschung dar. Die geometrische Struktur der Oberflächen kann auf atomarer Skala mit verschiedenen Mikroskopiemethoden im Realraum abgebildet werden. Die genaue atomare Struktur wird mittels Informationen aus Elektronenspektroskopie, dem Rastertunnelmikroskop und der Beugung langsamer Elektronen bestimmt. Einige Beispiele aus dem Spektrum solcher Untersuchungen werden vorgestellt.
Äußere und innere Grenzflächen von Festkörpern
Ein wichtiger Aspekt der modernen Festkörperforschung ist die Herstellung und Untersuchung neuartiger Materialien mit maßgeschneiderten Eigenschaften. Dabei liegen aktuelle Zielrichtungen in der Herstellung von mehrkomponentigen Materialien, der Miniaturisierung bis zur Funktionalität auf der Nanometerskala, oder in der Herstellung von strukturierten Multischichtsystemen. Eine wichtige Funktion kommt bei der Entwicklung solch neuer Materialien der Kontrolle ihrer Grenzflächen und dünnster Materialschichten zu. Bei der Untersuchung dieser Grenzflächen und Schichten müssen deren chemische Zusammensetzung, die atomare Struktur und die elektronischen Eigenschaften ermittelt werden. Die Oberfläche des Festkörpers ist seine wichtigste Grenzfläche, da sie seine Kontaktstelle zur Umgebung darstellt, sei es für einen elektrischen Kontakt, für eine chemische Reaktion oder eine mechanische Anbindung. Bei immer kleiner werdenden Strukturen in den Materialproben kann die Oberfläche sogar die physikalischen Eigenschaften des Systems dominieren. So befinden sich in einem Würfel von 10 nm Seitenlänge bereits fast 7% der Atome direkt an einer Oberfläche, bei 1 nm Seitenlänge sind es etwa die Hälfte. Aber auch die inneren Grenzflächen beeinflussen entscheidend die Eigenschaften eines Festkörpers. Die Grenzfläche zwischen zwei unterschiedlichen Materialschichten bestimmt die elektronischen oder magnetischen Eigenschaften. Atomare Diffusion zwischen Schichten wird entscheidend durch die chemische Zusammensetzung an der Grenzfläche beeinflusst. Beim Aufbringen einer neuen Schicht während der Herstellung einer Probe wird die Oberfläche zur neuen inneren Grenzfläche. Sie bestimmt zunächst das Wachstumsverhalten und danach die physikalischen Eigenschaften des Systems. Am Max-Planck-Institut für Festkörperforschung wird die Untersuchung dieser vielschichtigen Fragestellungen seit kurzem in der wissenschaftlichen Servicegruppe für Grenzflächenanalytik betrieben.
Realraumabbildung
 Terrassen und Stufen einer 4H-SiC(0001)-Oberfläche nach Behandlung durch H2-Ätzen, aufgenommen mit dem AFM. (b),(c) Mikroskopie auf atomarer Skala mit dem STM: Terrassen mit unterschiedlicher atomarer Anordnung (b) auf 4H-SiC(0001) nach Si-Bedampfung und Heizen im UHV, geordnete Adatome (c) der (3×3)-Phase auf 4H-SiC(0001). Die Bildgrößen sind jeweils angegeben.")

Ein erster Zugang zur Charakterisierung der Oberfläche einer Festkörperprobe ist die Bestimmung ihrer Morphologie, d.h. der geometrischen Struktur der Oberfläche. Dies beinhaltet einerseits die Ermittlung von Stufenhöhen und Terrassengrößen sowie eine Abschätzung von deren Regelmäßigkeit. Andererseits ist die Verteilung und Beschaffenheit von Defekten von Interesse. Mit dem atomaren Kraftmikroskop (AFM für atomic force microscopy) können solche Untersuchungen mit hoher Auflösung gemacht und Stufenhöhen bis in atomare Dimensionen bestimmt werden. Als Beispiel ist in Abbildung 1(a) die Oberfläche einer einkristallinen Siliziumkarbidprobe gezeigt, die mit dem AFM aufgenommen wurde. Durch geeignete thermische Behandlung in einem H2-Gasstrom wurde vorher gezielt SiC-Material abgetragen (dies wird auch als Wasserstoffätzen bezeichnet). Nach der Behandlung besitzt die Oberfläche atomar glatte Terrassen mit einer Stufenhöhe von ca. 1 nm, was genau einer Einheitsmasche der vertikalen Kristallstruktur der untersuchten 4H-SiC-Probe entspricht. Wie in dem Bild zu erkennen ist, sind die Terrassenbreiten im Bereich von ca. 0,5 µm angesiedelt. Dadurch lässt sich bestimmen, dass die Orientierung der Probenoberfläche nur ca. 0,1° von der Terrassenfläche, der so genannten SiC(0001)-Fläche, abweicht. In einer systematischen Untersuchung mit unterschiedlichen Ätztemperaturen konnte gezeigt werden, dass sich eine solch glatte Oberfläche nur bei einer bestimmten Temperatur (hier 1400°C) erzielen lässt. Bei höheren Temperaturen bündeln sich die Stufen zusammen, oder es entstehen gar tiefe Ätzgruben [1]. Ein ähnliches Verhalten wurde auch für anders orientierte SiC-Oberflächen gefunden [2].
 bzw. die Kupferkonzentration (rechts) innerhalb der mit AES zugänglichen Schichtdicke (wenige Atomlagen) wieder.")

Da die AFM-Aufnahmen in atmosphärischer Umgebung gewonnen wurden und die Oberfläche dadurch ständig von einem Feuchtigkeitsfilm belegt ist, können einzelne Atome nicht nachgewiesen werden. Im Ultrahochvakuum (UHV, bei einem Druck von ca. 10-10 hPa) dagegen lässt sich die Oberfläche des Kristalls unbeeinträchtigt von Verunreinigungen sauber präparieren. Unter solchen Bedingungen kann man mit dem Rastertunnelmikroskop (STM für scanning tunneling microscope) die Oberfläche mit atomarer Auflösung untersuchen. Dabei werden nicht die Atome selbst abgebildet, sondern es wird die Elektronendichte im Bereich der Oberfläche abgetastet, die ihrerseits mit der atomaren Struktur eng zusammenhängt. Abbildung 1(b) zeigt auf einer im UHV präparierten SiC(0001)-Oberfläche Terrassen mit Bereichen unterschiedlicher atomarer Anordnung [3]. Eine regelmäßige Anordnung der äußersten Atome auf SiC in einer (3×3)-Periodizität (dreifacher Atomabstand im Vergleich zur Volumenstruktur) ist in Abbildung 1(c) gezeigt. Man bezeichnet diese geordnete Oberflächenphase mit der Nomenklatur (3×3)-SiC(0001). Wie weiter unten gezeigt, können derartige Aufnahmen als erster Schritt zu einer Bestimmung der atomaren Struktur der Oberfläche dienen [4].
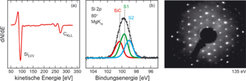
-SiC(0001)-Phase: (a) AES-Spektrum, Auftragung des differenziellen Signals dN/dE gegen die kinetische Energie der Elektronen. (b) Hochaufgelöstes XPS-Spektrum der Si 2p Linie mit chemisch getrennten Komponenten aus der Linienformanalyse. (c) LEED-Bild bei senkrechter Elektroneneinfallsgeometrie und Primärenergie der Elektronen von 135 eV.")
Für bestimmte Anwendungen ist es von Interesse, die Atome der Oberfläche auch chemisch zu identifizieren. Dies lässt sich mit dem Raster-Auger-Mikroskop (SAM für scanning Auger microscope) erzielen, indem die Probe mit einem feinfokussierten Elektronenstrahl abgerastert wird. Dabei werden Augerelektronen emittiert, deren kinetische Energie elementspezifisch ist und mit einem energieempfindlichen Spektrometer nachgewiesen wird. So lässt sich eine chemische Abbildung der Oberfläche gewinnen, wie in Abbildung 2 gezeigt. Auch mit dem STM lässt sich eine chemisch differenzierte Abbildung erzielen, in diesem Falle aber indirekt über lokale Unterschiede in der elektronischen Struktur.
Atomare Struktur der Oberfläche
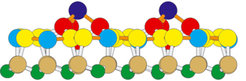
-SiC(0001)-Phase in Seitenansicht. Eine Identifikation der verschiedenen Atome (Kugeln) ist im Text gegeben.")
Für ein detailliertes Verständnis der Eigenschaften einer Festkörperoberfläche ist eine Analyse ihrer atomaren Struktur unumgänglich. Für die heute untersuchten komplexen Materialsysteme ist hierzu eine Kombination verschiedener Untersuchungsmethoden notwendig. Neben der oben erwähnten Abbildung im Realraum mit dem STM verwendet man Elektronenspektroskopie zur chemischen Analyse und Beugungsmethoden zur Kristallographie. Die Elementzusammensetzung im Oberflächenbereich wird durch Analyse der Bindungsenergien von Rumpfniveauelektronen ermittelt. Dies kann einerseits durch direkten Nachweis der Elektronenemission aus einer inneren Elektronenschale des Atoms nach Röntgenbestrahlung geschehen, der so genannten Röntgenphotoelektronenspektroskopie (XPS für X-ray photoelectron spectroscopy). Alternativ steht hierfür die Augerelektronenspektroskopie (AES) zur Verfügung, bei der die Elektronenemission nach dem Füllen einer solchen Schale nach der Ionisation beobachtet wird. In beiden Fällen ist die kinetische Energie des emittierten Elektrons ein Fingerabdruck für das jeweilige Element. Eine genauere Betrachtung der Linienform in den Emissionsspektren erlaubt zusätzlich, Aussagen über die chemische Umgebung des betreffenden Atoms zu machen. Da die Elektronen nur aus einer sehr begrenzten Schicht (wenige Ångstrøm Tiefe) aus der Oberfläche austreten können, sind die beiden Messmethoden sehr oberflächenempfindlich. Für die oben bereits erwähnte (3×3)-SiC(0001)-Phase sind in Abbildung 3 ein AES- (a) und ein XPS-Spektrum (b) gezeigt. Die Energieposition der Siliziumlinie in AES (LVV bezeichnet die beteiligten Elektronenschalen) zeigt, dass die Si-Atome an der Oberfläche direkt zu weiteren Si-Atomen gebunden sind, im Gegensatz zum SiC-Volumenkristall, wo es immer vier Kohlenstoffatome sind [5]. Der Kohlenstoffpeak dagegen zeigt die typische Karbidbindung des Volumenkristalls. In der hochaufgelösten Darstellung der Si 2p Emissionslinie im XPS in Abbildung 3(b) sind drei verschiedene durch eine Linienformanalyse ermittelte Komponenten eingezeichnet, die Si mit jeweils unterschiedlichen Bindungsnachbarn repräsentieren [5].
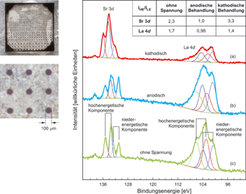
. Die grün gezeichnete Kurve (c) wurde an einer Elektrode gemessen, die lediglich thermisch aber ohne elektrisches Potenzial behandelt wurde. Die blaue und rote Kurve repräsentieren Elektroden nach anodischer (b) und kathodischer (a) Behandlung (siehe [7] für weitere Details). Links sind elektronenmikroskopische Aufnahmen der mikrostrukturierten Probe gezeigt.")
Mithilfe der Vorkenntnisse aus Mikroskopie und Spektroskopie über die Oberflächenphase kann nun eine komplette kristallographische Analyse durchgeführt werden. Dazu verwendet man die Beugung langsamer Elektronen (LEED für Low-Energy Electron Diffraction). Abbildung 3(c) zeigt ein Beugungsbild der (3×3)-SiC(0001)-Phase. Neben den hellen Beugungsreflexen der ersten Ordnung der Substratperiodizität sieht man eine Fülle zusätzlicher, so genannter Überstrukturreflexe auf einem Gitter der dreifachen Dichte. Die Intensitäten all dieser Reflexe werden bei veränderlicher kinetischer Energie der Elektronen vermessen und mit denen aus Modellrechnungen gewonnenen verglichen. Dazu wird zunächst ein atomares Modell der Oberflächenstruktur aus den oben erwähnten Vorkenntnissen entwickelt. Durch gezielte Veränderungen der atomaren Positionen wird die Übereinstimmung zwischen gemessenen Intensitäten und denen der Modellrechnung verbessert, bis eine ausreichende Strukturoptimierung gelungen ist [6]. Aus einer solchen LEED-Strukturanalyse konnte die Oberflächengeometrie der (3×3)-SiC(0001)-Phase ermittelt werden [4]. Abbildung 4 zeigt das optimierte Oberflächenmodell in einer Seitenansicht (die Oberfläche zeigt nach oben). Die obersten, dunkelblauen Si-Atome sind diejenigen, die in der STM-Aufnahme in Abbildung 1(c) zu sehen sind. Sie definieren die (3×3)-Periodizität, die sowohl im STM als auch in LEED zu sehen ist. Diese so genannten Adatome werden jeweils von drei weiteren, rot gezeichneten Si-Atomen „getragen“, die ihrerseits auf einer flachen, vollständig gefüllten Siliziumlage (gelbe und hellblaue Atome) liegen. Erst darunter befindet sich die erste volumenartige SiC-Bilage mit den hellbraunen Si-Atomen oben und den kleiner gezeichneten grünen Kohlenstoffatomen darunter. Diese Lage wird nach unten durch den SiC-Volumenkristall fortgesetzt (nicht gezeichnet).
Chemische Analyse und Tiefenprofile
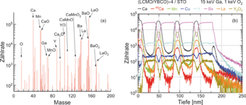
 Massenspektrum im Bereich einer Grenzfläche, (b) Tiefenprofil der verschiedenen Konstituenten der Probe durch das gesamte Schichtsystem. Die Zählraten der Ionen sind logarithmisch aufgetragen.")
Verschiedene spektroskopische Methoden werden zur Analyse komplexerer Proben und tieferer Schichten verwendet. Mit der oben bereits beschriebenen Elektronenspektroskopie kann die chemische Zusammensetzung an der Oberfläche und eine eventuelle Veränderung bei einer funktionellen Behandlung bestimmt werden. Mit der heute zur Verfügung stehenden Instrumentierung ist dies auch auf kleinen Nachweisbereichen möglich. Mit dem XPS können Bereiche bis hinunter zu wenigen Mikrometern Ausdehnung analysiert werden. Das SAM erlaubt sogar Auflösungen bis in den Bereich weniger Nanometer. Auf einer Testprobe mit La0,6Sr0,4Co0,8Fe0,2O3-δ als Basismaterial für Brennstoffzellen wurden Mikroelektroden mit 100 µm Durchmesser und kleiner elektrolytisch behandelt. Elektronenmikroskopische Aufnahmen der Probe sind in Abbildung 5 (links) gezeigt. Die Zusammensetzung der Oberflächenschicht als Funktion der Behandlung wurde mit XPS untersucht. Abbildung 5 zeigt rechts eine Linienprofilanalyse der Sr 3d und La 4d Emissionslinie. Für beide Elemente zeigt sich ein erheblicher Einfluss der Polarität der angelegten Spannung auf die XPS-Intensität, und damit auf ihren Anteil in der Oberflächenschicht nach der Behandlung. Durch die Profilanalyse können jeweils zwei unterschiedliche chemische Konfigurationen für Sr und La differenziert werden (die Linien haben selbst jeweils zwei Komponenten auf Grund der so genannten Spin-Bahn-Aufspaltung). Im Falle des Sr repräsentiert die Komponente bei etwas höherer Bindungsenergie eine Position in der obersten Oxidlage. Damit kann die aus der Abbildung ersichtliche Erhöhung der Sr-Konzentration durch die kathodische Behandlung auf eine Erhöhung dieser oberflächenspezifischen Komponente zurückgeführt werden [7]. Im Falle des La ist eine derartige Differenzierung nicht vorhanden.
. Die simulierte Stickstoffkonzentration ist durch die rote Kurve dargestellt und durch die linke und obere Achse gegeben.")
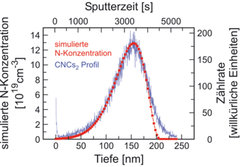
Eine solche chemische Analyse kann auch in die Tiefe der Probe fortgesetzt werden, indem die oberen Schichten durch einen hochenergetischen Ionenstrahl abgetragen werden (so genanntes „Sputtern“). Damit kann ein Tiefenprofil der Zusammensetzung erstellt werden. Dies wird während der XPS-Experimente ebenso durchgeführt wie während einer Analyse mit dem SAM. Mit diesen beiden Methoden ist der nachweisbaren Konzentration allerdings eine untere Grenze im Bereich von 0,1-1% gesetzt. Wesentlich genauer geht dies mit der Sekundärionen-Massenspektroskopie (SIMS). Eine besonders vielseitige Variante dieser Methode weist die Sekundärionen durch eine Flugzeitmessung (TOF für time-of-flight) nach. Diese Methode wird daher als TOF-SIMS abgekürzt. Durch einen (ersten) hochenergetischen Ionenstrahl werden die Sekundärionen zunächst erzeugt und im Flugzeitspektrometer nachgewiesen. Mit einem zweiten Ionenstrahl wird simultan (im regelmäßigen Wechsel) ein Krater in der Probe erzeugt. Durch Speichern der Ionenintensitäten verschiedener Massen können so Tiefenprofile aufgenommen werden. Damit können z.B. Schichtzusammensetzung und Grenzflächenbeschaffenheit in einem Multischichtsystem untersucht werden. Abbildung 6 zeigt dies an einer Probe aus sich abwechselnden La2/3Ca1/3MnO3 und YBa2Cu3O7-Schichten auf einem SrTiO3- Substrat [8]. Abbildung 6(a) zeigt ein Massenspektrum, das im Bereich eines Schichtübergangs aufgenommen wurde. Von vielen, teilweise zusammengesetzten Ionen werden diejenigen gewählt, die einen hohen dynamischen Bereich für die Zusammensetzung versprechen, und deren Intensität für die gesamte Tiefe analysiert. Ein solches Tiefenprofil ist in Abbildung 6(b) gezeigt. Zusammensetzung und Grenzflächenschärfe sind in einem weiten Größenordnungsbereich gut zu beobachten. Gleichzeitig erkennt man aber auch, dass die Interpretation der Messdaten durch variierende Nachweisfaktoren bei unterschiedlicher Filmstruktur (variierenden Ionisationswahrscheinlichkeiten) und durch eine Durchmischung beim Sputterprozess limitiert ist [8].
Mit hervorragender quantitativer Auflösung kann TOF-SIMS verwendet werden, um die Diffusion von Minoritätselementen, z.B. in Oxiden, oder die Konzentration von Dotierstoffen in Halbleitern zu analysieren. Hier ist eine konzentrationsunabhängige Eichung durch eine Referenzprobe möglich. Abbildung 7 zeigt als eine solche Referenz das Konzentrationsprofil von Stickstoff in einer SiC-Probe nach der Implantation von ca. 1,2·1020. Teilchen pro cm3. Im aufsteigenden Bereich wird die simulierte Konzentration exakt durch die TOF-SIMS-Messung wiedergegeben. Im weiteren Bereich stört dann die Durchmischung durch den Sputterprozess. Mit der so geeichten Nachweiswahrscheinlichkeit kann die Stickstoffkonzentration in SiC-Halbleiterschichten bis in den Bereich von 5·1016 cm-3 bestimmt werden [9].